



吴斌教授团队发现NF-κBp65-Ser536为肝癌关键药物靶点
2021年8月11日,Journal of Experimental & Clinical Cancer Research杂志在线发表拉斯维加斯lswjs0567官方网站吴斌教授团队题为“Phosphorylation of NF-κBp65 drives inflammation-mediated hepatocellular carcinogenesis and is a novel therapeutic target”的长篇研究论著。

图1 2021年8月11日J EXP CLIN CANC RES在线发表的论著全文
该研究发现肝脏慢性炎症反应,尤其是TNF-α,诱发肝细胞NF-κBp65表达上调,进一步通过β-arrestin1介导NF-κBp65-Ser536位点磷酸化,继而激活下游Akt/mTOR信号通路,促进了肝癌的发生与发展。该研究是吴斌教授团队在Nature Communications发表题为“β-arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling”及在Autophagy发表题为“HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G1/S cycle”研究工作的延续及深入。
该团队在人肝癌组织样本及血液样本中检测发现NF-κB信号通路5个亚基NF-κB1、NF-κB2、p65(RelA)、RelB、c-Rel中尤以p65(RelA)的表达及磷酸化最显著。根据这个重要临床发现,该团队进一步通过肝细胞特异敲除(L- NF-κBp65 KO)小鼠构建肝癌模型,与野生型小鼠比较,L-NF-κBp65基因敲除后肝癌发生的数量、体积等均显著低于野生型小鼠。在DEN、四氯化碳、TNF-α构建的3种小鼠肝脏炎症模型中,NF-κBp65的表达水平及磷酸化均显著升高;肝癌细胞实验及裸鼠成瘤实验中,NF-κB信号通路抑制剂(Bay 11-7082、PDTC)明显抑制肝癌细胞的增殖及移植瘤的生长。进一步通过β-arrestin1基因敲除小鼠、肝癌细胞β-arrestin1基因沉默实验、及NF-κBp65磷酸化位点突变实验等进行深入的机制研究,发现β-arrestin1显著激活NF-κBp65-Ser536位点的磷酸化,但并不激活Ser276或Ser529位点的磷酸化,而Ser536位点的磷酸化活化了下游Akt/mTOR信号通路,结果诱发肝细胞的癌变及促进肝癌的发展。
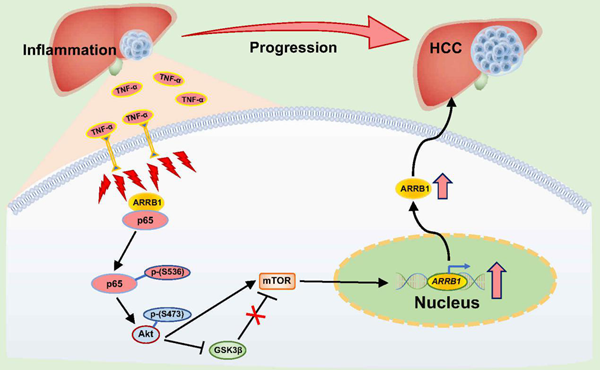
图2 在肝脏炎症微环境中,TNF-α激活NF-κB信号通路,在β-arrestin1(ARRB1)诱导下NF-κBp65亚基Ser536位点磷酸化上调,从而活化下游Akt/mTOR信号通路,以诱发肝细胞的癌变及促进肝癌的发展
此项研究为理解肝脏炎症环境中NF-κBp65的显著上调及磷酸化在肝癌中的作用及相关机制提供了新的科学依据,同时揭示了β-arrestin1诱导NF-κBp65-Ser536磷酸化的新机制,为NF-κBp65及其Ser536位点磷酸化作为肝癌防治的关键药物靶点提供了新的理论基础。
吴斌教授的学生徐璇博士与雷一鸣博士为此文的共同第一作者,吴斌教授和杨逸冬副主任医师为共同通讯作者,拉斯维加斯lswjs0567官方网站为论文第一作者单位及通讯作者单位。该研究获得国家自然科学重点项目及面上项目(U1501224与82070574)、广东省自然科学基金团队项目(2018B030312009)的支持。
论文链接:https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-02062-x

